Li-11-3
Developmental
disorders of Bile Ducts
By
Dr. E. Orfei
CONTENTS
Cysts
Congenital polycystic disease
Von Meyenburg complex
Congenital hepatic fibrosis
DILATATIONS
Bile duct atresia, intrahepatic and extrahepatic, syndromatic and non-syndromatic
Caroli's syndrome
Byler's syndrome
Alagille Syndrome
Aagenaes syndrome
Benign
recurrent intrahepatic cholestasis
North american
indian cholestasis
Recurrent
benign cholestasis of pregnancy
SIMPLE CYSTS
They can be solitary or multiple, from few mm to 20 cm in diameter,
unilocular. They arise from aberrant bile ducts. They are lined by a uniform
cuboidal or columnar epithelium resembling bile duct epithelium. They contain
clear fluid. Large cysts are seen almost exclusively in women. They are usually
aymptomatic. Complications are rare and my consist of intracystic bleeding,
rupture, purulent infection, compression of inferior vena cava, compression of
common bile duct at its bifurcation with resulting jaundice, compression of
portal vein with resulting portal hypertension, communication with an
intrahepatic bile duct, torsion, carcinoma. Simple cysts of the liver, especially
the solitary ones, are not associated with renal cysts. If they are
multiple, they are sometimes part of the adult cystic disease involving the kidneys.
The diagnosis
is made with ultrasonography. Other imaging techniques are less effective.
Differential diagnosis
includes:
hydatid cyst, liver abscess, hemangioma, hematoma, cystadenoma, cystic necrotic
malignant tumor, adult polycystic kidney disease when the cyst in the liver are
multiple. Adult polycystic renal disease is always associated with simple
multiple cysts of the liver but not vice versa
CONGENITAL
POLYCYSTIC
LIVER
Polycystic disease affects mostly the kidneys but it may involve also the
liver, lungs, spleen and pancreas. The liver is involved in the following renal
cystic patterns.
1-Adult polycystic renal disease
(hereditary
autosomal dominant). It is a congenital cystic disease of long duration. It
starts to give symptoms in the 3rd and 4th decade of adult life. It causes
progressive renal failure and death in around the 5th and 6th decades due to
progressive cyst formation replacing the renal parenchyma. About 40 % of
patients have multiple cysts of the liver. Presence of cysts in the kidneys may
be seen already in the fetus and in the neonate. Their presence in the liver is
seen in the 20s and 30s and their frequency will progressively increase with the
age. The disease is hereditary autosomal dominant with the gene localized in the
short arm of chromosome 16. The transmission is therefore penetrant and the
frequency is 1:500 to 1:1000 throughout the world.
2-Childhood polycystic renal disease
(hereditary
autosomal recessive). This cystic disease is seen early in life, in the fetus,
in the newborn, in the infant, in the juvenile and also in early adult age. The
disease may be so severe in the fetus
that it will be incompatible with life. In the newborn
the renal cysts may be mild and the liver may show only fibrosis. In
adolescents the renal lesions can be
seen with ultrasonography as in adult polycystic kidney.
The liver will be affected by diffuse fibrosis which will cause portal
hypertension. Younger children die of severe renal disease, Older children die
of severe hepatic fibrosis and complications of portal hypertesion.
The disease is hereditary but autosomal recessive. The carrier parents are
not affected and only 25% of children of both carrier parents may be affected by
this disorder.
3-
Medullary sponge
kidney.
In these kidneys the cortex is normal but the collecting tubules of the medulla are dilated
and contain urine and calculi. The changes are bilateral. The affected
individuals, 1:5000, develop no renal failure but nephrolithiasis. And becomes
symptomatic around the 5th decade. The diagnosis is made by intravenous urography.
The liver may demonstrate congenital hepatic fibrosis and Caroli’s disease.
4-Medullary cystic renal disease associated with congenital malformations.
This disorder consists of medullary cysts of various sizes in the
cortico-medullary junction of the kidney and is associated by other congenital
malformations. The most frequent associations are those of brain malformations
such as raquischisis, occipital meningoencephalocele, polydactilia, abnormalities
of the heart and eyes and congenital hepatic fibrosis which compose the Meckel
syndrome. Another syndrome includes Dandy-Walker malformation( cystic dilatation
of the fourth ventricle and absence of the cerebellar vermis). These disorders are
incompatible with life.
In all above conditions the cysts in the liver vary in size. They are lined
by columnar, cuboidal or flat bile duct epitheluim. The fibrous stroma contains
often Von Meyenburg complexes.
In the liver the cysts are usually clinically silent unless there are
complications, such as : pressure on bile ducts causing jaundice, pressure on
portal vein causing portal hypertension , infection. The diagnosis is made with
ultrasonography and CT scan. Prognosis of polycystic liver depends on the age of
the patient and the degree of renal involvement. In adults the prognosis is good
because liver cysts rarely compromise live functions. Death in these patients is
caused by renal failure due to concomitant polycystic kidneys. In children ,
especially in the perinatal period the disease may be fatal because of the
marked renal involvement.
Click
on the pictures to enlarge
|
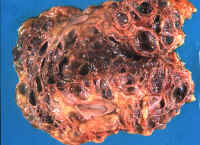
Fig.
li.11-3-1 Adult polycystic disease
Both kidneys are transformed into cystic masses weighing 600grams
each. The patient was a 59 year old female.
The cysts are large and had caused chronic renal failure. The patient was
kept alive for over two years with renal dialysis. The liver was moderately involved by the cystic disease.
|
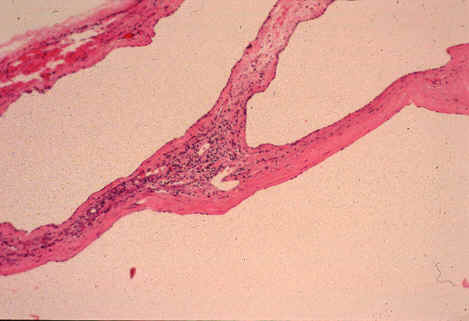
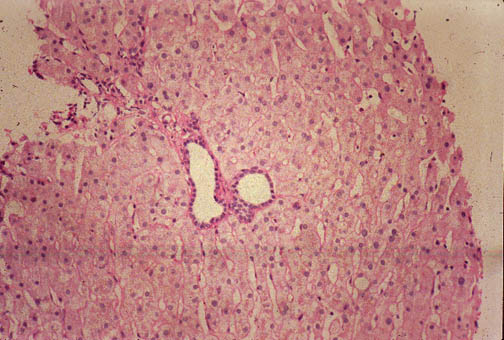
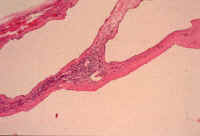 Fig. li-11-3-2. Microscopic view of the cysts.
The cysts are lined bile duct epithelium which, in these cysts, it is cuboidal. There are
also multiple Meyenburg complexes.
Fig. li-11-3-2. Microscopic view of the cysts.
The cysts are lined bile duct epithelium which, in these cysts, it is cuboidal. There are
also multiple Meyenburg complexes.
|
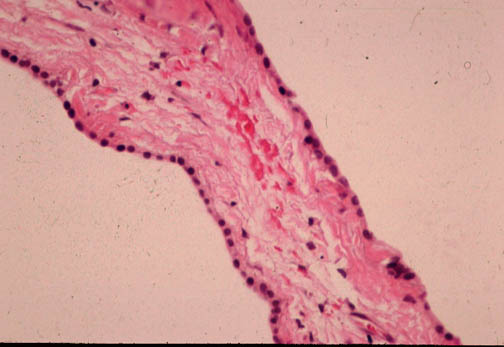
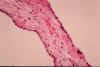 Fig. li-11-3-2A. Cuboidal epithelium of cyts wall.
Fig. li-11-3-2A. Cuboidal epithelium of cyts wall.
|
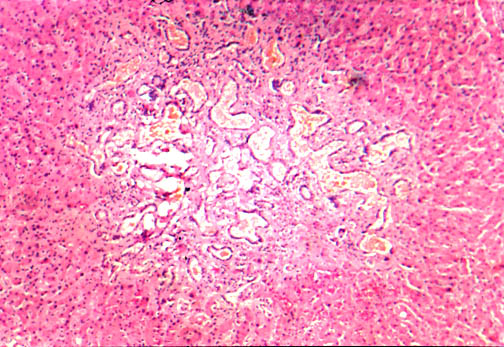
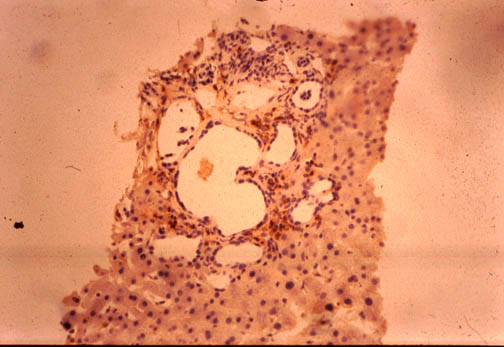
 Fig. li-11-3-3. Von Meyenburg Complex.
Fig. li-11-3-3. Von Meyenburg Complex.
They are small
gray-white or green small nodules
consisting of numerous bile
ducts embedded in fibrous stroma, sometimes dilated an sometimes containing bile
stained material . Their location is periportal. They have been observed to
degenerate into adenomatous and adenocarcinomatous neoplasia.They are considered
part of the adult polycystic disease.
|
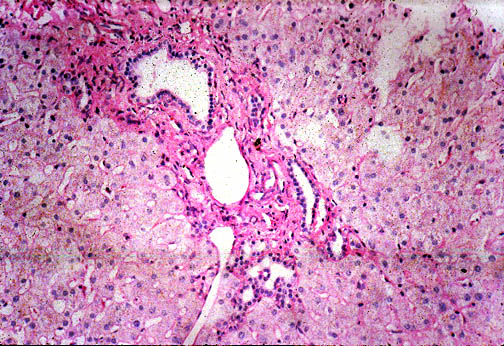
CONGENITAL HEPATIC FIBROSIS
It is characterized by expanding portal fibrosis containing proliferated and
dilated bile ducts communicating with the biliary tree. It is not a cystic
disease because the cysts do not communicate with the biliary tree. It is not
cirrhosis because there are no regenerative nodules and no active septa with
inflammatory cells. It is not Caroli's’ syndrome because in Coroli’s the
dilated bile ducts are not accompanied by aggressive fibrosis and there is no
portal hypertension. The initial basic disorder is probably proliferation and
dilatation of portal bile ducts. The fibrosis is secondary. In this disorder the
dilated ducts do not form large cysts like in polycystic disease. The disorder
is inherited as autosomal recessive trait. The frequency is 1:10,000.
Clinically
it causes portal hypertension which my be present at birth but it is recognized after years when it has produce complications of portal hypertension, especially
bleeding esophageal varices.
Since the dilated bile ducts communicate with the biliary tree they can get
infected by bacteria and develop acute ascending cholangitis similar to Caroli’s
disease which is dilatation and proliferation of portal bile ducts but without
fibrosis and without portal hypertension. The patient with congenital hepatic
fibrosis die of complications of portal hypertension and rarely of cholangitis;
the patients with Caroli’s disease die of ascending cholangitis.
Congenital hepatic fibrosis may be associated with polycystic disease
intestinal lymphangioectasia, cleft palate, aneurysms of cerebral and renal
arteries, pulmonary emphysema and cerebellar hemangioma
.
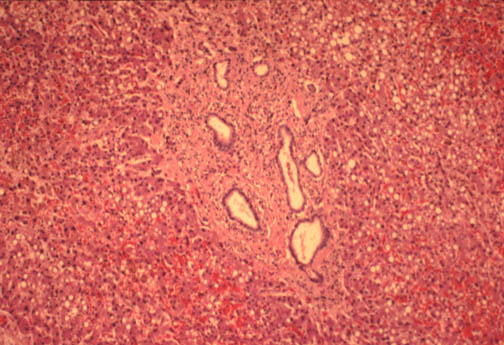
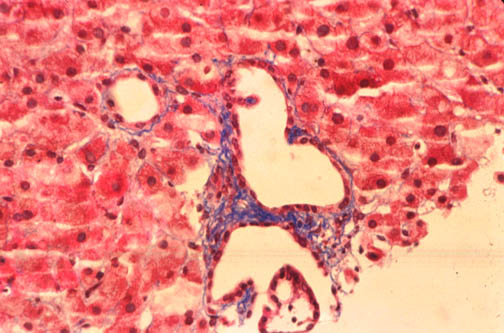
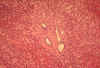 Fig.
li-11-3-4. Congenital
hepatic fibrosis. Fig.
li-11-3-4. Congenital
hepatic fibrosis.
The
portal field is expanded by fibrosis a contains
numerous dilated interlobular bile ducts.
Notice the sharp demarcation between portal
fibrosis and surrounding liver parechyma. There is no inflammatory reaction.
|

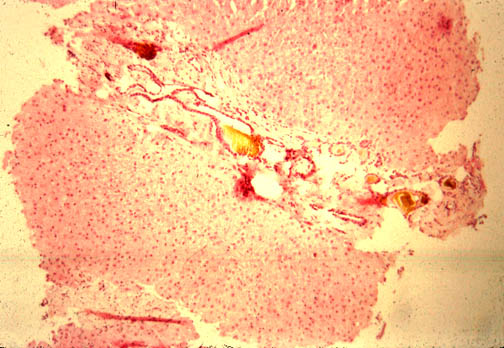
Fig.
li-11-3-5. Congenital hepatic fibrosis.
The
parenchyma does not show any fibrous dissection nor regenerative nodules.
|
CONGENITAL DILATATIONS OF BILE DUCTS
Developmental dilatation disorders involve extra and intra hepatic bile
ducts.
Type I - Dilatation
of common bile duct
Type II- Diverticulum of common bile duct and gallbladder
Type III-Choledococele
Type IV-Multiple intrahepatic dilatations (Caroli’s
syndrome)
Type V -Localized dilatations of major intrahepatic bile ducts
Caroli’s syndrome
It consists of multiple dilatations
of portal bile ducts without aggressive fibrosis, at difference
with congenital hepatic fibrosis. The syndrome, therefore, does not cause portal hypertension.
The complication in Caroli’s
is infection: acute ascending cholangitis. Caroli’s changes may be associated
in the majority of cases with congenital hepatic fibrosis. The associated form
with congenital fibrosis is inherited as an autosomal recessive trait same as
congenital hepatic fibrosis. The form not associated with congenital
hepatic fibrosis is not inherited.
Strumental investigation of the biliary tree in both conditions should be
avoided as much as possible because i
Click
on the pictures for enlargement
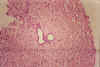 Fig.
li-11-3-6. Fragment of
needle biopsy. Portal bile ducts are dilated and empty. There is no
portal fibrosis. The dilated ducts
are commnicating with yhe biliary system. They may, therefore, contain
bileand may contain purulent exudate of an ascending cholangitis. They
do not cause portal hypertension because do not produces porta
fibrosis. Fig.
li-11-3-6. Fragment of
needle biopsy. Portal bile ducts are dilated and empty. There is no
portal fibrosis. The dilated ducts
are commnicating with yhe biliary system. They may, therefore, contain
bileand may contain purulent exudate of an ascending cholangitis. They
do not cause portal hypertension because do not produces porta
fibrosis.
|
.
Fig.
li-11-3-7.
The
dilated ducts are lined by cuboidal epithelium. |
 Fig.
li-11-3-8. Another case of Caroli's yndrome diagnosed by needle
biopsy. Notice in this case stained for collagen the absence of portal
fibrosis which is present in congenital hepatic fibrosis where there are
also dilated bile ducts. Fig.
li-11-3-8. Another case of Caroli's yndrome diagnosed by needle
biopsy. Notice in this case stained for collagen the absence of portal
fibrosis which is present in congenital hepatic fibrosis where there are
also dilated bile ducts. |
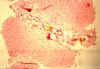 Fig.
li-11-3-9. Fig.
li-11-3-9.
In this case
the ducts contain inspissated bile. There is no fibrosis. The bile
containt confirms the open communicating nature of these dilated channels. |
|
 Fig.
li-11-3-10. Fig.
li-11-3-10.
In
this case you see dilated ducts and portal fibrosis. There is a
combination of congenital ductal dilatation (Caroli' syndrome) and
congenital hepatic fibrosis. |
BILIARY ATRESIA
Biliary atresia is the obliteration of the lumen of the bile ducts. It may
involve extrahepatic and intrahepatic ducts, separately or together.
Extrahepatic bile duct atresia is seen in 1:10000 live births and is present
at birth. It is suspected when the icterus in a neonate appears in 2-3 weeks
after birth and does not resolve. It is important to diagnose this condition in
the first 10 weeks of life because the process tends to spread to the
intrahepatic ducts and the defect from initially correctable will become non-correctable
by Moiro Kasai operation of porto-enterostomy.
It is therefore imperative to intervene with corrective surgery as soon as
possible, within the fist 10 weeks of life. Without surgery, the life span is
1-2 years. Surgery restores bile flow in about 50% of the cases but about 40% of
the cases will develop post surgical complications which are cholangitis and
less frequently portal hypertension because of progressive loss of bile ducts
and development of cirrhosis. Five year survival after surgery is around 36%.
Correctable Correctable Correctable Non- Correctable
Gross pathology
The alteration may involve segments which are resectable and an anastomosis
between dudenum and the remaining ducts at the porta hepatis can be made. In
other cases the obliteration is more extensive involving the upper branches of
the bile ducts and a similar anastomosis cannot be performed. The operation was
devised by a Japanese surgeon, Moiro Kasai.
Microscopic pathology
(Gautier
m, Eliot N, Arch Path & Lab Med 105:397, 1981)
1)- Complete fibrous obliteration of the ductal lumen without any evidence o
ductal epithelium.
2)-Ducts with altered lumina with coboidal epithelium and periluminal
neutrophilic infiltration.
3)-Ducts with altered lumina and incomplete epithelium.




The intrahepatic bile duct atresia is never complete.
There are almost always some interlobular bile duct in some portal fields.
It is therefore more appropriate to talk of
scarcity of intrahepatic bile
ducts or better in latin term:
PAUCITY
of intrahepatic bile ducts. The condition is diagnosed if the
interlobular bile duct/portal field ratio is 0.5 or less.
The normal ratio is between 0.9 and 1.8. In order to make this
evaluation it is needed to have a biopsy with a sufficient number of portal fields, at
least 6. It is therefore preferable to have a wedge biopsy rather than a needle biopsy
of the liver. It is also necessary to use the best stains to visualize the
portal bile ducts such as
immunostains
for keratins
specific for bile duct epithelium.
The scarcity of intrahepatic bile ducts in neonates may be an isolated
(non-syndromatic
form) or it may be accompanied with other
developmental defects (syndromatic
form).
Non- syndromatic biliary atresia.
Beyler’s Syndrome.
Paucity of intrahepatic bile ducts since birth with progressive jaundice and
death in 1-2 years.
-Less common than syndromatic
paucity of bile ducts.
- worse prognosis than
syndromantic form.
Ethiology:Unknown. Idiopathic or
associated with: rubella, alpha-1-antitrypsin deficiency, Down’s syndrome, Turner
syndrome, trisomy17-18, treehydroxycoprostanic acid excess, congenital syphilis.
Pathology:
Paucity and progressive
disappearance of intrahepatic bile ducts with periportal fibrosis leading to
cirrhosis in 50% of cases and to hepatocellular carcinoma. Probably related to
adulthood ductopenia.
Clinically:
Jaundice in
early life with
high direct bilirubin, bile acids and alk. Phosphatase. Vit E, D, A deficiency
leading to neurological changes, rickets and lickenified skin.
Inheritance:
autosomal recessive form.
First reported in 1969 in an Amish family named Byler.
North
american indian cholestasis.
This
condition was described in 1981 in 14 North American Indian children from
several families by Weber (Gstroenterology 81:653,1981). It
consists of jaundice, hepatomegaly and facial telangioectasia. The disease
progresses to cirrhosis and death. Toward the end, the jaundice frequently
disappears. Histologically the findings are those of neonatal
hepatitis in very young children; later are those of post hepatitic
cirrhosis. There is marked increase of pericanalicular microphilaments similar
to phalloidin poisonong. The cause, therefore appears to be adysfunction
of the microphilaments.
Benign
recurrent intrahepatic cholestasis.
This
condition was described in Lancet in 1959 by Summerskill, WHJ and Walshe
JM (Lancet 2: 686, 1959).
It
consists of recurrent attacks of pruritus and jaundice with clear intervals
characterized by asence of signs of hepatic dysfunction. The first attack
usually starts in chldhood before buberty but it may not occurr before the third
decade of life.
It
stars with intense pruritus and macular eruptions for one-two weeks and the by
pruritus and jaundice in qui severe forms thathat my persist for a short time as
two weks and for a long time as two years. The average duration is 3-4 months.
Fall is the most common season. The symptom-free intervals are of variable
duretion ranging from few months to several years. During the attack
there is increase of serum bilibin, mostly
conjugated even above 20 mg/100ml, inrease of bile acids and alkaline
phosphatase. The liver bipsy sows centrolobular cholestasi and moderate
mononuclear infiltration of the portal area. During free intervals the hepatic
functions are normal and the liver is morphologically normal. The syndrome may
be familial. The conditio is totally benign without long term sequelae.
(Familial)
recurrent intrahepatic cholestasis of pregnancy.
This
condtion cosista of insurgens of pruritus with or without moderate jaundice
during the last trimester of pregnancy.
The
form without jaundice is called "pruritus gravidarum". The syndrome
promptly disappears post partum. It is attributed to the marked increase of
placental and gonadal hormones characteristic of pregnancy. Such patients are
also liable to develep jaundice with oral contraceptives. The condition may be
familial. It is bebign to the mother but not benign to the fetus by inceasing
maternal morbidity. Histologically the liver shows simple cholestasis with
centrolobula intracytoplamic and canalicular retention of bile pigment without
significant inflammation.
Syndromatic biliary atresia.
Aagenaes
syndrome.
Tis
condition was recognized In Norway by Aagenaes et al. in 1968 (Archives of
Diseases in Childhood 43:646.1968)
It
is also called: Norwegian cholestasis an cholestasis with lymphoedema. It
consists of jaundice in the neonatal period
which
may clear and recurr intermittently during life. The jaundice is
accompnied with marked edema of the lower extremities. The edema persists during
the period free of jaundice. The liver biopsy may show paucity of intrahepatic
bile ducts, ductular proliferation and giant cell hepatitis. In adults the
syndrome may cause cirrhosis.
Alagille Syndrome
The syndromatic form of this congenital disorder is more common and has
better prognosis than the non-syndromatic form. It was reported for the first
time by Alagille in 1975 in a patient with " hepatic-ductular hypoplasia,
characteristic facies, vertebral malformations, retarded physical, mental, and
sexual development and cardiac murmur. Journal of Pediatrics, 86: 63-71, 1975".
Clinical:
Juandice in infancy and
childhood resolving with age, elevated serum direct bilirubin, bile acids, alk
phosphatase. Def of Vit D, E, K with rickets, neuropathy and coagulopathy.
Pathology:
paucity of intrahepatic bile
ducts which are lost with advancing age. Loose periductal fibrosis in disappearing
bile ducts. Reduction in the number of portal fields per 10 square millimeters of liver tissue
(8.2 in normal, 4.5 in biliary atresia). This fact indicates a defect of
embryological ramification of the biliary tract.
Similarly it has been found by angiography a defective ramification of the
pulmonary arteries. The basic defect in the liver appears therefore to be a
defect of embryological ramification of the portal vessels.
TO
CONTENTS/ BILIARY