li-11-1
BILIRUBIN
METABOLISM By
Dr. E. Orfei
Physiology and
pathology
1-Bilirubin
production.
2-Transport in
blood.
3-Hepatocellular
uptake.
4-Intracellular
transport in hepatocytes.
5-Conjugation with
glucuronic acid.
6-Secretion into
bile ducts.
7- Intestinal
metabolism.
8- Renal excretion
of bilirubin
9- Renal excretion
oh urobilinogen
1-BILIRUBIN
PRODUCTION
Bilirubin is the
terminal product of heme metabolism. Heme is present in hemoglobin and in other
oxidative compounds such as hepatic mitochondrial and microsomal cytochromes
(P-450). Thus plasma bilirubin is part erythropoietic and part non-erythropoietic.
Approximately, 85 % erythropoietic and 15% non-erythropoietic.
The erythropoietic
fraction originates from two sources: the circulating normal aging red cells and
the immature defective red cells of the bone marrow. The daily production of
bilirubin is 250 to 350 mg.
Shunt bilirubin is
called that portion that does not originate from senescent circulating red cells
but originates from immature and defective red cells (7%) and from non- hemoglobin
heme compounds, particularly from hepatic cytochromes and from myoglobin. These
two fractions were discovered by labeling hemoglobin with a radioactive glycin,
and observing that one fraction (78 %) of bilirubin is excreted in the feces in
120 days and another fraction is excreted in 10 days or less. The first was
called late labeled bilirubin, the second was called early
labeled bilirubin or shunt bilirubin. Shunt bilirubin may be markedly
elevated in certain pathologic states: sideroblastic anemia, megaloblastic
anemia, erythroleukemia, lead poisoning and a congenital disorder called "idiopathic
dyserythropoietic jaundice". The patients affected by this condition do
not have hemolysis. They have hyperbilirubinemia and jaundice. The
hyprbilirubinemia is due to shunt bilirubin.
Bilirubin from
erythropoietic heme is produced by monocytic macrophages, reticulo-endothelium,
in every organ but especially in the spleen, liver and bone marrow in order of
importance.. The bilirubin from non-erythropoietic hepatic heme is produced in
the hepatocytes.
The tetrapyrrolic
ring of heme is broken by an oxygenase at the alpha bridge, the bond
between the two carbons opposite to the gamma bridge which is between the two
carbons carrying the two propionic acids. The tetrapyrrolic molecule from a ring
is transformed into a tetrapyrrolic chain without iron.
HEME
+ Heme
oxygenase = OXY- HEME
( closed tetrapyrrolic ring with iron)
OXY- HEME
+ heme reductase = BILIVERDIN
(open tetrapyrrolic ring without iron)
BILIVERDIN
+ biliverdin reductase = BILIRUBIN
(unconjugated)
Pathology
of bilirubin production
Hyprbilirubinimia
with jaundice occurs in increased destruction of red blood cells namely: hemolysis.
It occurs in 1)congenital disorders of red cells (sickle cells, thalassemia,
spherocytosis), 2) immune hemolysis (erythroblastosis fetalis, 3) acquired
diseases of red cells (dyserythropoiesis), etc.
In the adult,
even a marked hemolysis does not produce significant increase of serum bilirubin
if the hepatic bilirubin clearance is normal. In the newborn, however, a
marked hemolysis will be catastrophic. At levels of 20mg/dl of serum
bilirubin the infant will be deeply jaundiced and will develop kernicterus (Nuclear
jaundice: a grave form of yellow staining and degeneration of intracranial gray matter especially of lenticular nucleus, ammon,s horn and subthalamic area).
Phototherapy
is used for treatment of hyerbilirubinemia in neonates.
Bilirubin is a photoreceptor.
The blue light transforms bilirubin into colorless products of oxidation which are excreted in the urine.
Synthetic
porphyrins containing tin or zinc instead of iron cause decrease of
bilirubin formation by competing for the heme oxygenase activity of macrophages.
These compounds have been used in the treatment of hyperbilirubinemia in animals
and humans (e.g. Gilberts syndrome) with limited success.
2-BILIRUBIN
TRANSPORT IN BLOOD
Bilirubin is toxic
to tissues, therefore, it is transported in the blood bound to albumin. Only a
minute amount of free form is present in the blood.
Pathology
of bilirubin transport in blood.
If the free
fraction increases, bilirubin will invade and damage the tissues. It will cross
the blood -brain barrier and cause kernicterus in the neonate. Free
plasma bilirubin can increase in the fallowing pathologic conditions:
-1-
overproduction.
-2- defective conjugation in the hepatocyte.
-3- presence of
substances interfering with bilirubin-albumin binding: sulphonamides , long-chain
fatty acids from breast milk, salycilates, contrast media, etc. These agents
compete for albumin binding sites.
3-HEPATOCELLULAR
UPTAKE OF BILIRUBIN.
Bilirubin is taken
up by hepatocytes at their sinusoidal surface. The albumin-bilirubin bond is broken. Albumin remains in the plasma. The free molecule of bilirubin enters
the hepatocyte.This uptake is very rapid.
Pathology
of bilirubin uptake by hepatocytes.
The impairment of
uptake will result in unconjugated hyperbilirubinemia.
Occurrence:
1) Male
fern oil jaundice. This oil was used to treat tape worm. (Aspidium).
3) Jegzichte
sheep.
4-INTRACELLULAR
TRANSPORT OF BILIRUBIN IN HEPATOCYTES.
In the hepatocye
bilirubin is bound to cytoplasmic proteins: ligandins and Z
protein. Ligandins are a group of enzymes that represent 2% of cytosolic
proteins. Z proteins bind fatty acids. The primary function of these proteins is
that of avoiding the reflux of free bilirubin into the blood. Apparently, the
time lapse between uptake of bilirubin and cojugation is relatively long.
Pathology
of intracellular transport.
No
hperbilirubinemia and jaundice is known due to deficiency of ligandins.
5-CONJUGATION
WITH GLUCURONIC ACID
One
way for cells to neutralize unwanted compounds is to conjugate them with a
modified sugar, a glycosyl. The sugars used for this reaction are xylose,
glucose or glucuronic acid. Glucose is normally present in the cell sap, xylose
and glucuronic acid are formed from glucose by UDP-glucose dehydrogenase.
Xylosidation is predominant in plants, glucosidation in bacteria
and glucuronidation in mammals. Unconjugated bilirubinin is lipophilic.
Its conjugation with glucuronic acid renders it hydrophilic, thus, it can be
eliminated in the bile. Many other agents are eliminated by conjugation with
glucuronic acid: steroids, thyroid hormone, catecholamines, estradiol,
testosterone, bile acids, phenols, morphine, which can be conjugated by other
cells besides hepatocytes.
The glucuronidation
of bile proceeds in two steps: first glucuronic aid (GA) is
synthesized from cytosolic glucose that is complexed with uridinediphosphate (UDP)
ad forms udpglucuronic acid (UDPGA). From this compound, the glucuronic
acid is transferred to blirubin. The first reaction is catalyzed by a UP-
glucose dehydrogenate, the second reaction is catalyzed by bilirubin-
DUGAN- transferees that is synthesized by microsomes. Any deficiency of
these two enzymes will result in defective conjugation and elimination of
bilirubin. On the other end, administration of microsomal enzyme inducers such
as phenobarbital, glutethimide and antipyrine favor bilirubin conjugation and
elimination by increasing blirubin transferase activity. Conjugation occurs in
the endoplasmic reticulum and consists of forming an ester between glucuronic
acid and one or both propionic side-chains of bilirubin. The result will be
formation of bilirubin mono and di-glucuronides. In general, about 80% of
the di and less than 20% of the mono are formed. Human bile cotains also small
amounts of unconjugated bilirubin. In summary:
GLUCOSE
+ UDP-Glucose-dehydrogenase = UDP-GLUCURONIC
ACID (UDPGA)
UDPGA
+ BILIRUBIN + Glucuronyl transferase = BILIRUBIN
MONO &
DI-
GLUCURONIDES.
Pathology
of bilirubin conjugation
GILBERT’SYNDROME
Is due to a very
mild deficiency of glucuronyl transferase.It is a very frequent disorder. It
affects 5 to 7% of the general population. More common in males. It consists of
mild fluctuating jaundice due to non- hemolytic unconjugated hyperbilirubinemia
in the range of 5 to 7mg/dl or rarely higher. The liver is morphologically
normal. State of health and life-span are normal. Hemolysis, low caloric diet,
nicotinic acid will increase the jaundice. A lipid diet will decrease the
jaundice. Phenobarbital and other enzyme inducing agents are beneficial. Some individuals
with this syndrome beside a defect of bilirubin
CRYGLER-NAJJAR
SYNDROME, TYPE I
Is due to a severe
deficiency of glucuronyl tranferase. Deep jaundice develops tat birth, High
serom unconjugated hyprbilirubinemia, >20 mg/dl., not responding to
phenobarbital. Absent formation of diglucuronides. Death usually in the first
year or two with kernicterus. Phototherapy, plasmaferesis and albumin exchange
are beneficial. Liver transplantation may be life-saving. The liver is
histologically normal. A similar condition exists in Gann rat. Fortunately this syndrome
is rare. Only 100 or more cases have been described. It is apparently a
hereditary autosomal recessive trait.
CRYGLER-
NAJJAR SYNDROME TYPE II
Is due to a
moderate deficiency of glucuronyl transferase. Milder unconjugated
hyperbilirubinemia responding to enzyme inducing agents: phenobarbital,
gltethimide, phenazone, chlorpromazine. Both, mono and di-glucuronides are
formed. Patients develop normally but some may suffer bilirubin encephalopathy,
kernicterus. They will have unremitting jaundice for the whole life. It is a
familial disorder. The mode of genetic transmission is not clear.Thi defect of conjugation
may have an associated defect of bilirubin uptake by hepatocytes.
PYSILOLOGICAL
JAUNDICE OF THE NEWBORN.
It is due to a
very transient insufficiency of glucuronyl transferase. During the first few
days of life there is an overproduction of bilirubin and an underdeveloped
mechanism of the liver to dispose of bilirubin.
Together with
deficient conjugation, bilirubin production, blood transport, hepatic uptake and
secretion are all deficient. Sometimes extrahepatic factors exist to aggravate
the situation: infections, drugs competing for binding sites of bilirubin and
above all, breast feeding. The long chains of fatty acids of the breast milk
interfere with bilirubin-albumin binding sites.
6-
BILE SECRETION FROM HEPATOCYTES
The liver is an
endocrine and an exocrine gland. It secretes synthesized products internally into
the blood through the sinusoidal surface such as blood proteins, coagulation
factors etc. and secretes external into the biliary tract and the intestine
bile and many other substances, the terminal products of detoxifying function.
The mechanism of this external secretion is the least clear in the physiology of
the liver. It seems that many cellular organelles are involved in this process:
vesicles, Golgi complexes, lysosomes, plasma membranes, mitochondria,
cytoskeleton, plasma membranes, canalicular villi. Are however clear the
consequences of the malfunction of this apparatus especially in the secretion
bile which will result in conjugated hyperbilirubinemia.
Pathology
of bile secretion
DUBIN-JOHNSON
SYNDROME.
The syndrome
consists of chronic benign
jaundice due to conjugated hyperbilirubinemia without pruritus
or
elevation of serum alkaline phosphatase nor histological evidence of cholestasis.
The hepatocytes contain an abundance of coarse
dark-brown pigment similar to melanin . The
liver is black but normal. Serum bilirubin ranges between 2 and 20mg/dl,
60% conjugated. Jaundice appears in the first 3 decades of life and is
intermittent. Sometimes the onset is acute, simulating a hepatitis. The prognosis
is excellent. The disease is inherited as autosomal
recessive trait. The diagnosis is made by
needle biopsy. Corriedale sheep have similar black liver disease.
Click
on the pictures to enlarge
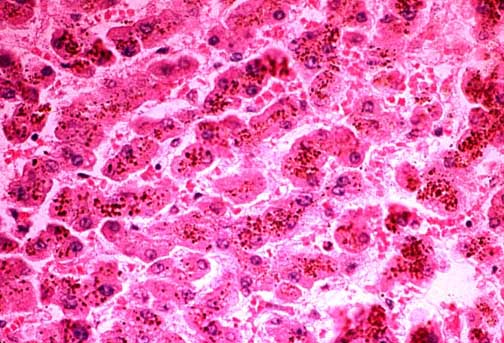
 Fig.11-1-1
Dubin-Johnson Syndrome. Fig.11-1-1
Dubin-Johnson Syndrome.
The
liver is brown-black because of the large amount of brownish coarse
pigment stored in the hepatocytes. Typically there is no intrahepatic
cholestasis in this condition. The pigment predominates in the
centrolobular zone. |
|
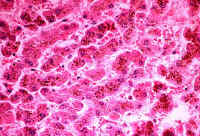 Fig.11-1-2
Dubin-Johnson Syndrome. Fig.11-1-2
Dubin-Johnson Syndrome.
There
could be a moderate portal fibrosis in older patients. The pigment is stored
in lysosomes like lipofuscin. Bile canaliculi do not contain bile. According
to studies conducted in Corriedale sheep, the pigment contains a
melanin-like component and and its formation is attributable to a
defect of excretion of epinephrine metabolites. |
ROTOR
SYNDROME.
This is a condition
similar to Dubin-Johnson. There is intermittent jaundice with conjugated
hyperbilirubinemia, similar clinical course, excellent prognosis but no
pigment in the liver tissue.
BENIGN
RECURRENT INTRAHEPATIC CHOLESTASIS.
A syndrome
characterized by recurrent attacks of rather severe jaundice. The attacks start
usually before puberty but they may start later. They are preceded by 2-4 weeks
of pruritus malaise, anorexia followed by
increasing
jaundice without pain or fever and lasting an average of 2-3 months during each
attack. It may last from tow weeks to two years.
Nausea, vomiting ,
abdominal pain and skin rash occur in some cases. An affected individual may have
up to 30 attacks during his life. Biochemically these patients have elevated
serum bilirubin, 10 to 20 mg/dl, mostly conjugated, elevated alkaline
phosphatase and bile acids. Alpa-glutamyl transferase (GGT) is elevated. Serum
bile acids are elevated 2-30 folds. Transaminases are occasionally markedly elevated. These abnormalities and the clinical
symptoms disappear completely in
disease-free intervals. In the cholestatic phase there is acinar zone 3
cholestasis with bile plugs and mononuclear cell infiltration in the cholestatic
area. In some cases there may be mild hepatocytic damage and portal mononuclear
infiltrate. These changes do not produce any fibrosis or cirrhosis. Liver
biopsies taken during clear intervals were normal. The disorder is rather rare
and appears to be familial with autosomal recessive character.
FAMILIAL
RECURRENT INTRAHEPATIC CHOLESTASIS OF PREGNANCY.
This disorder is clinically
and biochemically similar to benign intrahepatic cholestasis. It
occurs in the third trimester of pregnancy when the estrogen level is the highest
and disappears postpartum. The affected subjects appear to belong to families
with benign intrahepatic cholestasis trait. Gonadal steroid appear to ply a determining
role in the cause of this syndrome. Histology of the liver shows
centrolobular cholestasis similar to benign intrahepatic cholestasis. It is most
frequent in Scandinavia (1/100), Bolivia and Chile (1/10).The disorder is safe
for the mother but not for the fetus who will suffer premature births and
stillbirths due to placental infarcts. The mothers have higher incidence of
gallstones. Sometimes the disorder manifests itself only with presence of
pruritus without jaundice. (Pruritus gravidarum). The patients are not
severely ill as in fatty liver of pregnancy, hepatitis, obstructive jaundice.
DRUG-
INDUCED INTRAHEPATIC CHOLESTASIS.
Many drugs produce
cholestasis. The first cases reported were due to chlopromazine and synthetic
steroids now out of market (Nilavar). Synthetic oral contraceptives are high in
the list. They appeared to act on sensitivity base and affect only sensitive
individuals. Many appear to impair the secretory function of the hepatocytes. And
the list is increasing with the advent of new drugs. The liver in these cases
may show
marked and fatal
necrosis.
POST-OPERATIVE
INTRAHEPATIC CHOLESTASIS
It is attributable
to the combined effect of bilirubin overload deriving from blood transfusions
and to defect of hepatocytic secretory function. Usually the jaundice appears in
1-2 postoperative days and disappears in one or two weeks, Hyperbilirubinemia is
predominantly conjugated with rater normal alkaline
phosphatase and
transaminases.
BACTERIAL
INFECTIONS
It is a form of
intrahepatic cholestasis. The hyperbilirubinemia is conjugated in all cases.
Elevation of serum alkaline phosphatase in some cases. Hepatic histology without
much hepatocellular damage.
Three types of
morphological changes have been described:
1-canalicular
cholestasis , the most common, mostly pericentral without hepatocellular
damage.
2-ductular
cholestasis, characterized by the presence of big bile thrombi in bile
ductules and canals of Hering at the periphery of portal fields. No bile plugs
in interlobular bile ducts.
3-Toxic shock syndrome
due to infection with staphylococcus aureus producing Toxic
Shock Syndrome Toxin-1 (TSST-1). This toxin was produced by this organism
growing in polyacrylate tampons in menstruating women. The liver suffers
inflammation of intrahepatic bile ducts and canaliculi.
with rupture of
bile ducts and microvesicular steatosis. There is inflammatory reaction in
portal fields with
neutrophils,
eosinophils lymphocytes and monocytes. There is centrolobular cholestasis in 50%
of cases.
7-
INTESTINAL METABOLISM OF BILIRUBIN
Bilirubin in the
intestine is reduced to urobilins according to the following cascade:
BILIRUBIN
GLUCURONIDE
+ bacterial
or intestinal beta-glucuronidase = FREE BILIRUBIN
FREE BILIRUBIN
+ bacterial dehydrogenase = UROBILINOGEN (colorless)
UROBILINOGEN +
dehydrogenase = UROBILIN (orange-yellow).
The bulk of
bilirubin, urobilinogen and urobilin is excreted in the feces. Small amounts of
bilirubin and urobilinogen are reabsorbed by the intestine and return to the
liver. The bilirubin is recunjugated in the liver and re-excreted in the feces.
The reabsorbed urobilinogen is excreted in the urine, about 4 mg/ day and 0,1 to
1 mg in a random urine sample.
Pathology
of biliary excretion into the intestine
COMPLETE
BILIARY OBSTRUCTION.
The bile does not
reach the intestine therefore the feces are acholic. There is conjugated
hyperbilirubinemia and bilirubinuria. Urobilinogen is not formed in the
intestine and there is no urobilinogen in the urine. because since the bile does
not reach the intestine, urolinogen is not formed.
PARTIAL
BILIARY OBSTRUCTION.
Less bile reaches
the intestine. Urobilinogen is formed but in smaller amounts. There is less
conjugated hyperbilirubinemia, absent bilirubinuria and small amounts of
urobilinogen in the urine.
HEMOLYSIS.
Hemolysis causes unconjugated
hyperbilirubinemia. There is no bilirubinuria because unconjugated bilirubin
is not hydrophilic and cannot be excreted in the urine. There is increased urobilinogen in the urine because more bilrubin reaches the intestine and more
urobilinogen is formed an reabsorbed.
8-
RENAL EXCRETION OF BILIRUBIN
Only conjugated bilirubin (the direct fraction) is excreted in the urine when
its level in the plasma is increased above normal. It not present in the urine
of normal subjects and it is not eliminated in the urine in cases of
unconjugated (the indirect fraction) hyperbilirubinemia, such as in cases of
hemolysis.
Only
the small fraction of non-protein bound bilirubin in the plasma passes in the
urine. Some drugs and bile salts which compete for protein binding (salicylates,
sofosoxazole) increase The theshold of elimination depends on the degree of
protein binding which varies and its quantity in the urine does not have clinical
relevance.
Conjugated
bilirubin can be demonstrated in the proximal renal tubules.
9-RENAL
EXCRETION OF UROBILINOGEN
Urobilinogen is formed by bacteria in the small intestine and in the
colon.
It
is then reabsorbed by the small intestine and the colon and re-xcreted by the by
the liver into the intestine almost entirely. A very small amount is therefore
excreted into the urine: 0-4 mg/day. This amount will increase when more
urobilinogen is formed or when the liver is sick and unable to re-excrete it.
This amount will decrease when its formation in the intestine is decreased such
as in the case of complete bile duct obstruction when the bile cannot flow to
the intestine where urobilinogen is formed by the specific bacteria. The
urobilinogen formed by bacteria in the small intestine is re-absorbed
better than that formed in the colon.
CONTENTS/
TO BILIARY SYSTEM
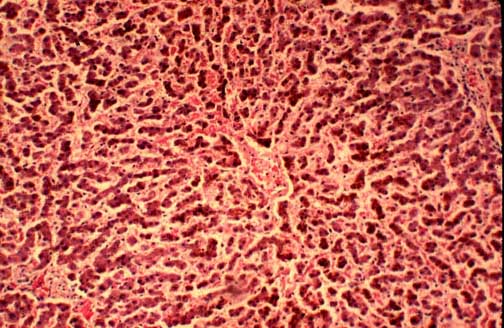 Fig.11-1-1
Dubin-Johnson Syndrome.
Fig.11-1-1
Dubin-Johnson Syndrome.