Fig.191- Cerebrotendineous xanthomatosis.
Fig.191- Cerebrotendineous xanthomatosis.
Section of cerebellum. The brain substance is infiltrated by masses of cholesterol clefts which are produced by cholesterol crystals dissolved by fat solvents during the histological preparation.
|
li-11-2 BILE ACIDS METABOLISM by Dr. E. Orfei
PHYSILOGY AND PATHOLOGY
Definition
Bile acids or, better, bile salts are the metabolites of cholesterol. Cholesterol metabolism occurs in the liver.
Formation from cholesterol
Bile acids are formed by removal of the last three carbon atoms of the terminal aliphatic side chain of cholesterol. It is thus formed a cyclopentanophenantrene molecule of 19 carbon atoms with a side chain of a variable number of carbon atoms. The side chain terminates with a carboxyl and may have from 1 to 8 carbon atoms. In humans the most common bile acids are those with a side chain of 5 carbon atoms and a total of 24 carbon atoms: the cholanoic acid. The enzyme operating the transformation of cholesterol into cholanoic acid is a microsomal enzyme, 7-alpha-hydroxylase.
Hydroxylation
By hydroxylating various carbon atoms of the molecule, more classes of bile acid are formed. The hydroxylation occurs in the liver and varies in different animal species. The major classes in man are:
-Monohydroxycholanoic acids. (Lithocholic, allolithocholic, isolithocholic).
-Dihydroxycholanoic acids. (Hyodeoxycholic, chenodeoxycholic, ursodeoxycholic, deoxycholic).
-Trihydroxycholanoic acids. (Cholic, Hyocholic, ursocholic, murocholic).
-Tetrahydroxycholanoic acids. (Phocaecholic, varanic acid).
-Triketocholanoic acids. (Dehydrocholic).
-Hydroxy-keto cholanoic acids. (Ketolithocholic).
Conjugation
Bile acids are conjugated with amino acids, glycine and taurine catalyzed by an acyltransferase. Detoxifying conjugation occurs also with sulphate, glucuronic acid (8%) and sugars. Sulphation and glucuronidation increase in cholestasis and cirrhosis. Taurine conjugates increase in cholestasis. Glycine conjugates increase in intestinal malabsorption because insufficient amounts of taurine is absorbed by the intestine. Taurine is an essential amino acid and it has to be obtained with the diet.
Secretion into the bile ducts.
Bile acid are actively secreted as salts into the bile canaliculus. The secretary activity depends on the availability of
cholesterol and phospholipids with which they form micelles.
Intestinal metabolism
The micelles of bile acids an cholesterol or phospholipids in the upper intestine are large and therefore are not reabsorbed but function as emulsifying agents in the digestion of lipids. In the lower intestine the micelles are smaller and are partially reabsorbed and are returned to the liver where they are reconjugated and re-excreted.
They are partially reabsorbed in the intestine and returned to the liver The portion that is not reabsorbed passes into the large bowel where they are deconjugated and metabolized by bacteria into secondary bile acids such as lithotomic acid. They are reabsorbed from the colon and returned to the liver but cannot be eliminated from the liver and passes into the blood. In normal conditions only a minor quantity of bile acids enters the systemic blood circulation. Their level increases very early in liver disease.
From the intestine they are eliminated in the feces. From the blood they are eliminated in the urine.
Pathology
Bile salts increase the elimination of water, cholesterol, lecithin and conjugated bilirubin in the bile.
In liver disease bile acid synthesis and turnover are decreased, therefore micelle formation and emulsification of fats is impaired , favoring the development of gall stones and steatorrhea.
Some defects of bile acids metabolism due to deficiency of some microtonal enzymes have been identified. All these defects cause homeostasis and some of them cause serious illnesses and death. The following syndromes have been reported.
-3-hydroxycoprostanic acid academia.
Severe homeostasis in two sibling infants who died at 8 and 20 months.(Hanson FRO et al:J Clin Invest 56:577,1975).
-Deficiency of 7-alpha hydroxylase leads to accumulation of monohydroxy bile acids (lithocholic and 3-hydrox- cholenoic) in blood and urine. The patient will suffer severe cholestasis. (Javitt NB et al: Gastroenterology and Nutrition. No.627: New York, Raven Press, 1986: 250A.)
-Deficiency of 3 beta hydroxy steroid dehydrogenase / delta isomerase.
Neonatal cholestasis and giant cell hepatitis (Cleyton PT et al. J Clin Invest 79:1031,1987)-Deficiency of Delta-3-oxosteroid-5 beta-reductase. Neonatal hepatitis in identical twins. (SetchellKDR et al.J Clin Invest 82:2148,1988).
-
Deficiency of 24 Hydroxylase. It is related to cerebrotendineous xanthomatosis and possibly to Zellweger syndrome.Cerebrotendineous xanthomatosis is a rare disease and consists of massive accumulations of chlesterol and cholesterol-like crystals in the brain and tendons causing tendon xanthomas, cataracts, cerebellar ataxia and dementia.
|
Fig.191- Cerebrotendineous xanthomatosis.
Section of cerebellum. The brain substance is infiltrated by masses of cholesterol clefts which are produced by cholesterol crystals dissolved by fat solvents during the histological preparation.
|
|
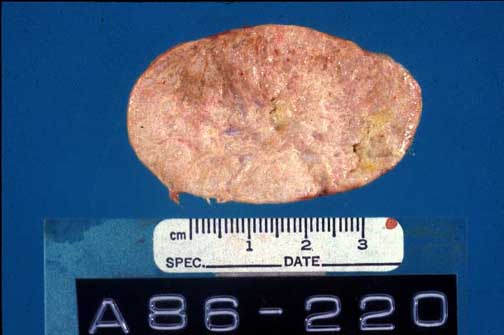
Gross section of Achilles tendon. Note the thickening and the yellowish discoloration of the tendon.
|
|
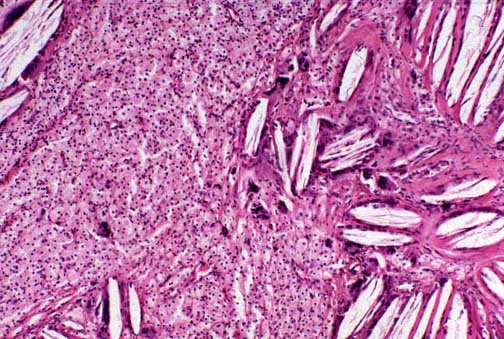
Achilles tendon. The microscopy shows massive infiltration of the tendon by cholesterol crystals.
|
The Zellweger syndrome, also called cerebro-hepato-renal syndrome is a fatal hereditary disease of infancy. It produces widespread gliosis of the brain, multiple small renal cysts and severe damage of the liver with necrosis, fibrosis, giant cells and hemosyderosis.The metabolic defect is not clear but there seems to be a block in the formation of chenodeoxycholic and cholic acid from cholesterol with accumulation of metabolic precursors of these compounds, dihydro, trihydro and tetrahydrocoprostanic acids, apparently toxic to the liver cells. There is absence of perixosomes and abnormalities of mitochondria in the cells of these individuals.