Huntington's Chorea / Huntington Disease
Physicians George Huntington (1872) and William Osler (1893) first described this hereditary form of chorea. Huntington Disease (HD) is a progressive disorder of motor, cognitive, and psychiatric disturbances.
The mean age of onset is 35 to 44 years (modified by repeat length, epigenetic influences, and possibly environmental influences) and the median survival time is 15 to 18 years after onset. Chorea, an involuntary movement disorder consisting of nonrepetitive, non-periodic jerking of limb, face, or trunk, is the major sign of the disease.
In the early stages, manifestations include subtle changes in coordination, minor involuntary movements, difficulty in mental planning, and often a depressed or irritable mood. In the next stage, chorea becomes more prominent with increasing difficulty with voluntary activity and worsening dysarthria and dysphagia. In late stages of HD, behavior problems are gradually lessened; motor disability becomes severe and the individual is often totally dependent, mute, and incontinent.
25% of individuals with HD have their first symptoms after age 50, some even after age 70, with a more benign course than typical. 5-10% of individuals with HD have juvenile HD with onset before age 21 years. Symptoms are similar but more severe, and seizures are common.
This disease occurs in 3-7 per 100,000 in populations of western European descent.
The HD gene is located at 4p16.3 (isolated in 1993), and the protein is named Huntingtin.
The disease is inherited in an autosomal dominant fashion, and is caused by the expansion of CAG trinucleotide repeats . New mutations are rare.
This disease is considered a disorder of inappropriate apoptosis, involving selective (localized) neural cell death. It is as yet unknown how the polyglutamine stretches created by the repeat expansions facilitate selective neuron degeneration.
Homozygotes for HD appear to have a similar age of onset to heterozygotes, but may exhibit an accelerated rate of disease progression.
Anticipation is the phenomenon in which increasing disease severity or decreasing age of onset is observed in successive generations. This occurs more often in paternal transmission of the mutated allele.
Diagnosis/Testing
Criteria for diagnosing an affected individual generally include the following:
- Family history consistent with autosomal dominant inheritance.*
- Progressive motor disability involving both involuntary and voluntary movement.
- Mental disturbances including cognitive declined and/or changes in personality
(Imaging studies can contribute additional support, but are mostly used to rule out other neurologic etiologies of the symptoms.) - Detection of a triplet-repeat (CAG) expansion in the HD gene (usually using PCR) (see table 1 for repeat categories).
[The Unified Huntington's Disease Rating Scale (HDRS) has been shown to provide a reliable and consistent assessment of the clinical features and progression of HD.] [Huntington Study Group 1996].
*Lack of a positive family history should not prevent a neurologist from considering this diagnosis. The asymptomatic father of an affected individual may have an intermediate allele, or either asymptomatic parent may have a reduced penetrance allele, or the disease may not have manifested itself in the parent yet (late onset).
Table 1 |
|
|
Allele Type |
Size (CAG repeats) |
Phenotype |
Normal |
< 26 |
Not symptomatic |
Intermediate |
27-35 |
Not symptomatic Males in this category are at risk of having a child with an allele in the mutant range. |
Reduced penetrance alleles |
36-39 |
May or may not develop symptoms of HD in their lifetime. |
Full penetrance alleles |
> 40 |
Huntington Disease |
Juvenile onset |
Usually > 60 |
Huntington Disease Usual onset < 21 years of age |
Predictive testing is used for adults in the absence of definite symptoms of the disease. (It is recommended to confirm a mutation in a known affected relative to be sure it is HD in the family before providing predictive testing). The chance that an at-risk person is affected decreases with age.
A standard multidisciplinary protocol, such as the HDSA Testing Guidelines [http://www.hdny.org/genetic_testing_guidelines.htm], are highly recommended. In fact, many labs won't accept samples without using a similar protocol. They should include:
- Pretest interviews in which the motives for requesting the test, the individual's knowledge of HD, the possible impact of positive and negative test results, and neurologic status are assessed.
- Counseling should include possible problems they may encounter with regard to health, life, and disability insurance coverage, employment and educational discrimination, and changes in social and family interaction.
- Consider implications for the at-risk status of other family members (parental status may be disclosed without consent)
- Informed consent should be obtained and records kept confidential.
- Individuals with a mutant allele need arrangements for long-term follow-up.
- See Genetic Testing: A Guide for Families at http://lkwdpl.org/hdsa/hdtest.htm#testing for a good patient-directed resource on these topics.
Predictive testing during childhood is consistently discouraged, but requests should be met with sensitive and understanding counseling. Arguments against this testing include taking away the child's right to decide whether or not they want to know; it raises the possibility of stigmatization within the family and in other social settings, and it could have serious educational and career implications. [See also the National Society of Genetic Counselors resolution on genetic testing of children and the American Society of Human Genetics and American College of Medical Genetics points to consider: ethical, legal, psychosocial implications of genetic testing in children and adolescents].
DNA banking (storing DNA) could be considered if there appear to be limitations to currently available testing for a specific family. Future improvements in testing methodologies may then be able to shed light on the recurrence risks and other issues.
Family planning The optimal time for determination of genetic risks and testing options is before a pregnancy has been conceived.
Prenatal testing can be performed for pregnancies at 50% risk by Chorionic villus sampling (CVS) or Amniocentesis. (The presence of an HD mutation in the family should be confirmed prior to achieving pregnancy.) This test is very rarely requested.
Ethical Issues for prenatal HD testing:
- Abortion of affected fetuses. Of course, abortion is a very complex issue in itself.
- Continuation of an affected fetus may:
- Violate the ideal of personal autonomy for the child. They are not consenting to this type of pre-symptomatic testing.
- Adversely affect upbringing, career choices, etc.
- Therefore, couples that would not terminate the pregnancy of an affected pregnancy are discouraged from this type of testing.
Pre-implantation Genetic Diagnosis is another option for those wanting to avoid passing on HD to the next generation. Diagnostic or exclusion testing could be performed on embryos after IVF, and only those either with a normal HD allele by diagnostic methods, or with a close to zero risk by exclusion testing, are implanted in the uterus.
Ethical Issues for PGD HD testing:
- Is an embryo a life? What is done with embryos that are not implanted?
Exclusion testing can be done prenatally by CVS or Amniocentesis, or Pre-implantation Genetic Diagnosis (PGD), using linkage analysis:
- This allows the parent-at-risk to remain unaware of their HD status, while preventing transmission of the disease to the next generation.
- This method does not diagnose the disease, but does trace which grandparent a chromosome was inherited from.
- The usual follow-up after this type of prenatal testing is to terminate a pregnancy with a ~50% risk and continue a pregnancy with a ~0% risk for HD. Using PGD, the embryos at ~50 risk would be implanted, and the ~0% risk embryos would not.
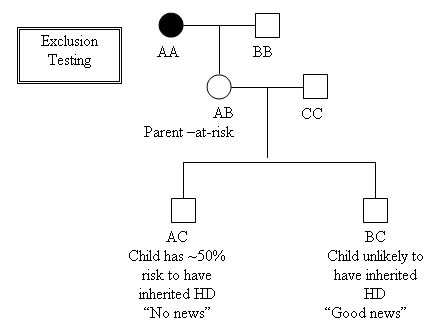
Ethical issues for Exclusion testing:
- Abortion
- This method would mean that approximately half of the abortions or non-implantations would be of non-affected fetuses/embryos.
- Continuing a pregnancy after knowing the fetus is at risk may have negative consequences for the fetus and the family (see ethical issues for prenatal HD testing).
Treatment
There is no cure for HD at this time. However, several areas of research are in the areas of neural and stem cell transplantation, apoptosis inhibitors, and inhibition of polyglutamine-induced protein aggregation.
Symptoms can be treated, including neuroleptics or benzodiazepines for chorea, and anti-parkinsonian agents for hypkinesia and rigidity, but L-dopa-containing compounds may increase chorea. Mental illness may be successfully treated by anti-depressants or anti-epileptic meds. There is no treatment for the cognitive impairment.
Supportive care with attention to nursing, diet, special equipment, and eligibility for state and federal benefits is very important for individuals with HD and their families. Local HD support groups can be extremely helpful.
Questions for students:
- Besides those listed above, what other options would a family have for not passing on Huntington disease to their descendants.
- List some reasons why a parent might want their child to have predictive testing.
- If a female patient presented to you with many of the clinical symptoms of HD, what further testing might you do to confirm the diagnosis? If she is affected, what are the main areas of support you can offer her?
- Her son wants predictive testing. What are the main issues he should be counseled about before having his blood drawn.
Resources
Huntington Disease Society of America (HDSA)
505 Eighth Ave.
New York , NY 10018
Ph: 800-345-HDSA (800-3345-4372); 212-242-1968
Fax: 212-239-3430
E-mail: hdsainfo@hdsa.org
www.hdsa.org
Lists testing centers and is an excellent resource for supporting patients and their families through testing, diagnosis and treatment.