Gauchers Disease
Gauchers disease is a treatable Lysosomal storage disease (figure 1). It is caused by the absence of the enzyme alpha glucsylceramide. There are many forms of this disorder. The most common is type I or nonneuropathic. Type I is characterized by hepatosplenomegaly, pulmonary disease, cytopenia and bone disease (Figure 2). Type II is also known as acute or infantile form. Clinical features include cognitive impairment, hepatosplenomegaly, cytopenia, pulmonary disease and dermatologic signs. Type II does not any bone changes. Type III is also known as subacute juvenile. Clinical features include hepatosplenogomegaly, cytopenia, pulmonary disease and seizures. The seizures are progressive.
Gaucher's disease is inherited in an autosomal recessive fashion. Type I is the most common type. It occurs in 1/815 individuals of Ashkenazi Jewish descent. Confirmation of this diagnosis can be made by enzyme analysis, DNA analysis or visualizing Gaucher cells on the bone marrow (Figure 3).
Evidence of bone disease is present in 70-100% of patients. Bone disease ranges from asymptomatic osteopenia to focal lytic or sclerotic lesions and osteonecrosis. Bone involvement may lead to acute or chronic bone pain, pathologic fractures, and subchondral joint collapse with secondary degenerative arthritis. Bone disease is often the most debilitating aspect of type 1 GD. The anemia universally present in GD has many causes including splenic sequestration. In advanced cases, decreased erythropoiesis as a result of bone marrow failure from Gaucher cell infiltration and medullary infarction.
The management of Gaucher disease involves symptomatic treatment of symptoms and enzyme replacement to treat the cause. Bone marrow transplant has also been shown to replace the ability to produce enzyme. Imiglucerase (Cerezyme ® ) is a recombinant glucosylceramidase enzyme . It is given via IV to patients with enzyme deficiency on a monthly basis. Regular intravenous infusions of imiglucerase have been demonstrated to be safe and effective in reversing those features resulting from hematologic and visceral (liver/spleen) involvement.
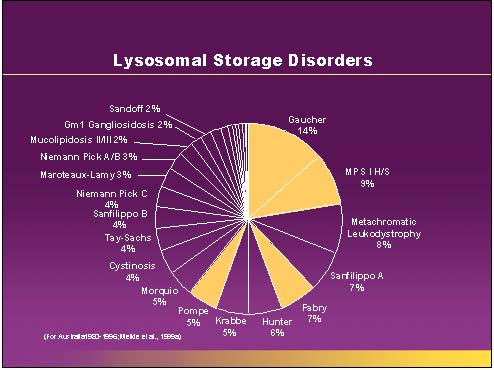
Figure 1.
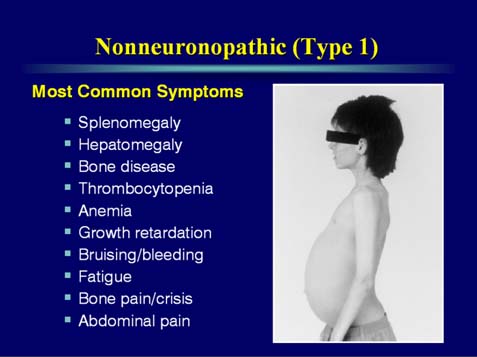
Figure 2.
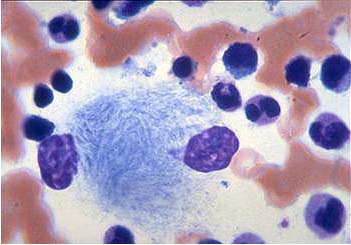
Figure 3.
A 4 year old boy is noted to have hepatosplenomegaly and anemia. He has normal cognitive development. He was born full term with no complications. The liver enlargement began 6 months earlier. The hepatosplenomegaly has been progressive over the past 6 months. He has recently begun complaining of leg pain.
- What would be the best method to diagnosis the cause of his problems?
- What is the recurrence risk for his future siblings also being affected?
- Management would include what treatment?