Cystic Fibrosis
Cystic Fibrosis (CF) is an inherited disorder that affects multiple systems. It is caused by mutations in the CFTR (cystic fibrosis transmembrane conductance regulator) gene, which is located on the long arm of chromosome 7. Mutations in CFTR also cause congenital bilateral absence of the vas deferens (CBAVD).
The diagnosis of CF is generally made by the presence of one or more of the characteristic features plus : the identification of two disease-causing mutations in the CFTR gene; OR two abnormal sweat chloride tests (which should be done at centers accredited by the CF Foundation) ; OR trans-epithelial nasal potential difference (NPD) measurements that are characteristic of CF. The diagnosis is now sometimes made by molecular genetic testing, such as through prenatal testing, when gene mutations have already been identified in the family.
The features of CF include:
- Pulmonary: chronic cough and sputum production, chronic wheeze, obstructive lung disease on lung function tests, pulmonary infections, chest x-ray abnormalities, digital clubbing. Pulmonary disease is the major cause of morbidity and mortality, with infections progressing to end-stage lung disease.
- Gastrointestinal/nutritional: Malabsorption/pancreatic insufficiency, intestinal obstruction, rectal prolapse, recurrent pancreatitis, meconium ileus, CF-related diabetes mellitus, chronic hepatobiliary disease, failure to thrive, fat-soluble vitamin deficiencies. Fifteen to twenty per cent of individuals with CF have meconium ileus at birth. The great majority (>90%) of patients have pancreatic insufficiency with malabsorption.
- Obstructive azoospermia; >95% of males are infertile as a result of azoospermia
- Salt-loss syndromes
The median survival is approximately 31-36 years of age. The disease can be highly variable, however, with some patients dying in early childhood as a result of lung disease and some only having recurrent sinusitis and bronchitis or male infertility in young adulthood. It is recommended that patients with CF be referred to regional CF centers, where they are seen by multidisciplinary teams.
CF is an autosomal recessive condition, meaning both parents are carriers of the disease. Couples with an affected child therefore have the following chances in a subsequent pregnancy: 25% chance of an unaffected child that is not a carrier; 50% chance of an unaffected carrier; and 25% chance of an affected child. CF is most common in the northern European populations; the disease incidence is one in 3,200 live births in Caucasians. The carrier frequency is ~1/22-1/29 in Caucasians, and lower in other ethnicities (Fig 1).
CFTR is the only gene known to be associated with CF. More than 1000 mutations in the gene have been identified; most, however, are rare and have only been detected in one family. ?F508 is by far the most common mutation; 66% of Caucasian carriers carry ?F508. Molecular genetic testing is available clinically. Laboratories will look for a panel of the most common mutations when doing mutation analysis. ACOG has recommended all women of child-bearing age be offered CF screening with a panel of 23 common mutations; some labs offer panels with a higher number of mutations. It is important to note that in population carrier screening, a negative CF screen result means the person's chance of being a carrier has been reduced but not eliminated (unless a CF mutation is already known to be in the family and is included on the panel). The amount of the risk reduction depends upon the person's ethnicity (Fig 1).
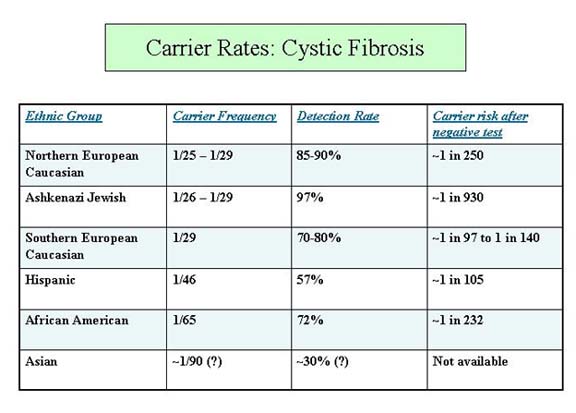
Figure 1.From March of Dimes/National Society of Genetic Counselors
A 4 year old Caucasian girl presents to clinic. She has failure to thrive, for which the etiology has not been determined. Her parents report she has chronic coughing and wheezing.
- What is her suspected diagnosis?
- What testing would you order?
- If your suspected diagnosis is confirmed, what are the chances her parents will have another affected child?